上一个分享,我们对测序数据进行了质量评估,接下来我们需要对数据进行数据质量控制。
数据预处理(Data Preprocessing):包括去除接头序列(adapter trimming)、去除低质量序列(quality filtering)、去除含有未知碱基的序列等,以净化原始测序数据。
进行质量控制的软件有fastp,Trimmomatic, trim_galore等,这里我们对这三个软件进行简要使用。
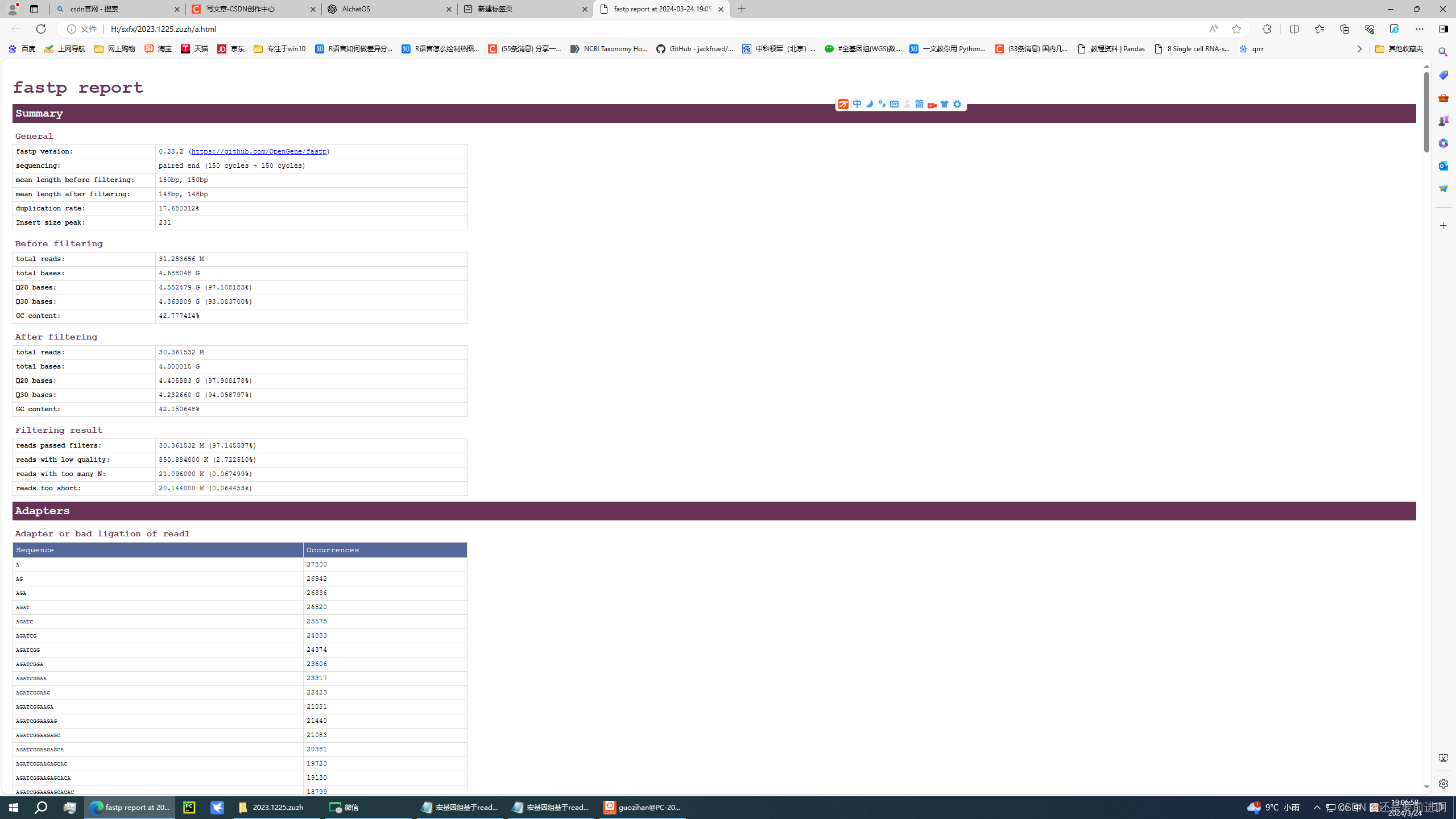
fastp的用法:以a1.fq.gz a2.fq.gz为例。
fastp --thread 4 -i ./a1.fq.gz -I ./a2.fq.gz -o ./a1.clean.fq.gz -O ./a2.clean.fq.gz -h L.html
fastp 是一个用于快速处理高通量测序数据的工具,主要用于质量控制和数据预处理。
-
-i, --in1:指定输入文件的路径,这通常是第一对(read1)原始测序数据的文件路径。
-
-I, --in2:指定第二个输入文件的路径,这通常是第二对(read2)原始测序数据的文件路径。
-
-o, --out1:指定第一个输出文件的路径,这是处理后的第一对测序数据(read1)的输出路径。
-
-O, --out2:指定第二个输出文件的路径,这是处理后的第二对测序数据(read2)的输出路径。
-
-h, --html:生成 HTML 格式的质量控制报告文件。
-
-j, --json:生成JSON格式的质量控制报告文件。
-
--thread:指定线程数,用于加速数据处理过程。
-
-q, --qualified_quality_phred:设定质量值阈值,低于该值的碱基将被截断。
-
-n, --low_complexity_filter:开启低复杂度序列过滤功能,用于去除包含低复杂度序列的 reads。
-
-e, --correction:开启错误校正功能,用于纠正测序错误。
-
-w, --overrepresentation_analysis:进行富集分析,检测PCR重复、接头污染等问题。
conda install -c bioconda Trimmomatic -y #安装 Trimmomatic,Trimmomatic是基于Java开发的,因此需要提前安装Java,才能使用Trimmomatic。
Trimmomatic -h #出现技术参数说明即可使用。
trimmomatic PE -phred33 -threads 4 a1.fq.gz a2.fq.gz a1_paired.fq.gz a1_unpaired.fq.gz a2_paired.fq.gz a2_unpaired.fq.gz LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36
PE表示处理双端测序数据。-phred33指定输入数据的 phred 分数编码为 phred33 格式。-threads 4指定了线程数为 4。a1.fq.gz和a2.fq.gz是输入的双端测序数据文件。a1_paired.fq.gz和a1_unpaired.fq.gz是第一个样本处理后的输出文件,分别包含配对的 reads 和未配对的 reads。a2_paired.fq.gz和a2_unpaired.fq.gz是第二个样本处理后的输出文件,分别包含配对的 reads 和未配对的 reads。LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36是指定的 trimmer 参数,用于指导 Trimmomatic 进行质量控制和去除接头序列等操作。
优势:
1、可使用参数更多,如滑窗剪切,可以直接选择使用内置的接头序列等等;
2、默认可生成paired和unpaired两种文件,更利于下游分析。
劣势:
1、代码非常长,而且容易写错,最好写在一个脚本里;
2、参数比较难记,像ILLUMINACLIP中的几个数字分别代表什么必须要对照说明书才能看懂;
3、运行时间较长;
4、只适用于illumina测序得到的数据,不适用于其他测序平台。
conda install -c bioconda trim_galore -y #需要python=3.7,创建一个新的环境指定python为3.7即可。
trim_galore --help #出现技术说明即可
trim_galore -q 20 --phred33 --length 36 -e 0.1 --stringency 3 --paired -o cleandata/ ./a1.fq.gz ./a2.fq.gz -j 15
-q 20:设置最低质量阈值为20,低于该值的碱基将被修剪。
--phred33:指定输入文件的质量值编码为Phred33格式。
--length 36:设置修剪后的读长为36,短于该长度的序列将被丢弃。
-e 0.1:设置错误率阈值为0.1,超过该阈值的序列将被丢弃。
--stringency 3:设置修剪的严格程度为3。
--paired:指示输入文件是成对的双端测序数据。
-o cleandata/:指定输出目录为"cleandata/",修剪后的结果文件将保存在该目录中。
./a1fq.gz ./a2.fq.gz:输入的成对测序数据文件。
-j 15:使用4个线程进行处理。










